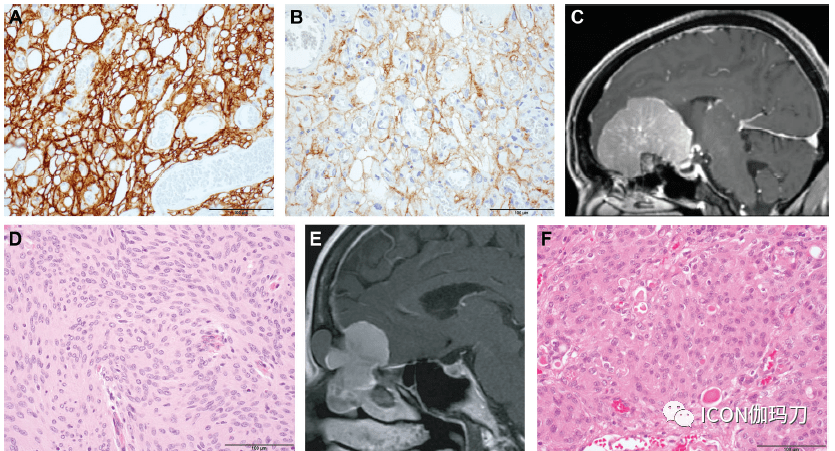
研讨停顿 神外前沿 神外前沿讯,近期,Neurosurgery杂志发表了美国Harvard Medical School的Wenya Linda Bi, Sandro Santagata撰写的综述《颅底肿瘤:神经病理学和临床意义》 论文信息: Skull Base Tumors: Neuropathology and Clinical implication doi: 10.1093/neuros/nyab209. 正文 来源自颅底和颅底周围的肿瘤包含普遍范围的常见和稀有的疾病实体。最近的研讨进步了我们对其发病机制的了解,在某些状况下,已显著影响临床理论。脑膜瘤的基因型与其表型(包含组织学亚型、肿瘤位置和临床结果)密切相关。 一个单一的分子改动,NAB2-STAT6融合,重新定义了孤立性纤维性肿瘤的类别,以包含以前的疾病实体血管外皮细胞瘤。神经鞘瘤,包含分发性的和家族性的,其特征是简直普遍存在的NF2的改动,在神经鞘瘤病中还有SMARCB1或LZTR1的突变。在垂体腺垂体肿瘤中,细胞谱系转录因子如SF-1、T-PIT和PIT-1往常是分类的关键,为以前被以为是零细胞腺瘤的肿瘤提供了更严厉的分类。垂体细胞谱系转录因子TTF-1定义了神经垂体肿瘤,它可能代表一个单一的病理实体,具有一系列的形态学表示(如颗粒细胞瘤、垂体细胞瘤和梭形细胞嗜酸细胞瘤)。 同样,脊索细胞谱系转录因子短链蛋白(brachyury)定义了脊索瘤,并将其与软骨肉瘤分辨开来。对成釉性颅咽管瘤和乳头状颅咽管瘤的非堆叠遗传驱动因子的审定表明,这些是不同的肿瘤实体,并已招致运用BRAF和/或丝裂原激活蛋白激酶抑止剂靶向治疗乳头状颅咽管瘤的胜利。相似地,在BRAF -突变和BRAF -野生型肿瘤的朗格汉斯细胞组织细胞增加症患者中也取得了显著的治疗反响。熟习颅底肿瘤的病理,其自然史和分子特征是优化患者医疗的必要条件。 颅底肿瘤是多种多样的成人和儿童肿瘤,来源于脑实质外,逾越不同的解剖距离,包含脑膜(如脑膜瘤、孤立纤维性肿瘤[SFTs])、颅神经(如神经鞘瘤)、鞍区(如垂体腺瘤、颗粒细胞瘤、颅咽管瘤)、骨及相关组织(如脊索瘤、软骨肉瘤)。 近年来,我们对许多颅底肿瘤的分子和遗传特征的了解有了很大的进步,从而进步了诊断、改善了预后,有时也改进了治疗计划。在此,我们对部分肿瘤实体的神经病理学提出简短的回想,突出与病人医疗相关的有创意的停顿。 脑膜瘤 脑膜瘤是一种典型的来源于蛛网膜帽细胞或其前体细胞的肿瘤的异质性群组,通常来源于颅底。这些肿瘤边疆分明,常伴有普遍的硬膜附着,在进袭性亚型(aggressive subtypes)中有侵袭颅底周围骨和软组织的倾向。生长抑素受体2a (SSTR2a)和上皮膜抗原(EMA)的免疫组化(IHC)可用于支持脑膜瘤的诊断,脑膜瘤最常用于不常见或稀有肿瘤的诊断(图1A和1B),NF2是50%以上脑膜瘤中最常见的突变基因。 脑膜瘤分为3个世界卫生组织(WHO)级别,反映不同的生物学和临床行为,至少有15种组织学亚型。约85%的脑膜瘤为1级,并遵照良性临床病程。假如有:(i)连续10个高倍镜视野(HPFs)下4 - 19个有丝团结象,(ii)小细胞间有超越2个不典型特征,核质比高(high nuclear-to-cytoplasmic ratio),细胞增加,核确判啮,连续的片状生长,如栓塞这样的干预性治疗所致的自发性坏死灶;(iii)明白的脑侵袭,肿瘤突起进入下面的GFAP阳性脑实质,而没有累及软脑膜(即,在Virchowo - Robin间隙中无血管周围扩散),或(iv)一种特定的形态学亚型(即脊索样和透明细胞),脑膜瘤被指定为2级(不典型)。固然目前运用脑部侵袭是不典型脑膜瘤的诊断规范,但队其价值仍需进一步研讨;固然在一些研讨中,脑部侵袭与相似于2级肿瘤的复发率增加有关,但其他研讨没有发现这种关联。 脑膜瘤假如有(i) 10个连续的高倍镜视野(HPF)下中有20个或以上的核团结象,或(ii)相似于肉瘤、癌或黑素瘤的明显间变性组织学特征,则归类为3级(间变性)。历史上,具有横纹肌样和乳头状形态(rhabdoid and papillary morphology)的肿瘤被划分为3级,但最近的研讨表明,横纹肌样脑膜瘤有不同的结果,应该依照上面所列的脑膜瘤的规范中止分级。 脑膜瘤的分级主要基于肿瘤组织学,分子遗传信息正在成为脑膜瘤样本特征和评价患者预后的关键(图2)。DNA甲基化方式和染色体缺失及增益负荷(burden of chromosomal losses and gains)与脑膜瘤复发风险增加相关。基因和甲基化标记物的特异性改动也与脑膜瘤的预后相关。TERT启动子的突变与肿瘤较高的分级和停顿、复发高风险增加的和较短的停顿距离有关。CDKN2A/B的缺失与复发风险的升高和停顿时间的缩短密切相关,而染色体上DMD基因的缺失也可能辨认出总体生存期较短的停顿性/较高级别脑膜瘤的一个亚群。BAP1或PBRM1的失活在进袭性横纹肌样和乳头状脑膜瘤中富集(enrich)。免疫组化察看到部分不典型和间变性脑膜瘤中H3K27me3缺失,与不良预后相关。 脑膜瘤基因型和表型(包含肿瘤位置、组织学亚型和行为)之间有很强的相关性。镰状脑膜瘤和矢状窦旁脑膜瘤在高级别病理中表示丰厚,与(发作在>50%的一切脑膜瘤中)NF2失活和染色体不稳定有关。相反,中线颅底脑膜瘤具有稳定的基因组,简直没有染色体的增加或丧失。来源于嗅沟或蝶窦平面的肿瘤大多为1级,通常为脑膜上皮型(图1C和1D), SMO或AKT1的激活突变富集。相比之下,分泌性脑膜瘤(图1E-1H)的特征是TRAF7和KLF4的突变。 这些假定的驱动基因(SMO、AKT1、PIK3CA、TRAF7和KLF4)的突变与更为常见的NF2突变或22q缺失(即22单体)相互排斥(mutually exclusive),而NF2和22q缺失在凸面和脊髓脑膜瘤中富集。经长期黄体酮治疗的女性脑膜瘤中PIK3CA突变丰厚,颅底位置也丰厚。此外,血管瘤和微囊组织可在同一脑膜瘤中同时发作,以取得多个染色体为特征(图1I和1J)。更为稀有的颅底脑膜瘤,如视神经鞘脑膜瘤,值得进一步的分子表征(图1K和1L)。这些基因组标签的停顿能够指导患者管理和临床实验的设计。
图1所示。脑膜瘤。脑膜瘤表示出A, SSTR2a和B, EMA的免疫反响性。C,T1加权增强矢状位MRI显现嗅沟脑膜瘤,伴有典型的脑膜上皮型组织学(H&E)。E,T1加权增强矢状MRI示嗅沟脑膜瘤延伸至蝶窦,显现F,分泌型组织学(H&E), G, PAS染色,H, CEA免疫组化证明。I, T1加权冠状位磁共振增强显现右侧前床突脑膜瘤伴J,混合血管瘤和微囊特征(H&E)。K, T1加权后斜矢状位磁共振检查视神经鞘脑膜瘤,并伴有L,脑膜上皮型组织学(H&E)。M,透明细胞型脑膜瘤(H&E)显现N,肿瘤细胞中SMARCE1免疫反响性丧失。CEA,癌胚抗原;EMA,上皮膜抗原;PAS,过碘酸-希夫染色反响;SSTR2a,生长抑素受体2a;比例尺,100 μm。 脑膜瘤的展开与遗传性肿瘤易理性综合征有关,最显著的是NF2。脑膜瘤的一切主要组织学亚型都发作在NF2患者中,但纤维母细胞亚型最为常见。NF2突变的严重水平与颅内和脊髓脑膜瘤的发作相关(即NF2外显子2-13的完整截断突变比错义突变或嵌合更严重)。许多NF2相关脑膜瘤是WHO 1级,但进袭性亚型也有发作。基因族群类型表型(Genotypephenotype)相关性也察看到在其他病症与透明细胞脑膜瘤脑膜瘤的风险增加的脊髓(这也能够发作在颅底)发作在生殖系突变SMARCE1(图1M和1N)和乳头状和横纹肌脑膜瘤的BAP1遗传突变。SMARCE1和BAP1的免疫组化用于辨认体细胞或体细胞或遗传背景下的蛋白表白的缺失。在有其他BAP1肿瘤易感综合征相关肿瘤,如葡萄膜黑色素瘤、间皮瘤、肾细胞癌和皮肤黑色素瘤,家族史的患者中,BAP1缺陷脑膜瘤的诊断应及时转诊中止种系检测。由于PTCH1或SUFU的种系突变招致的有痣样基内情胞癌综合征的家族中也有脑膜瘤的报道。
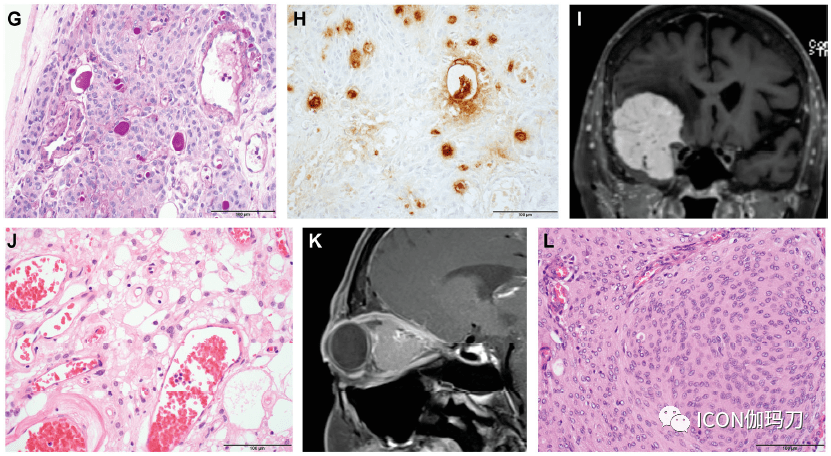
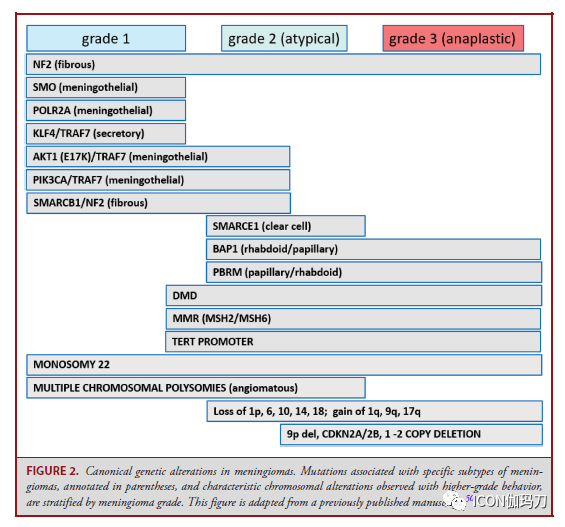
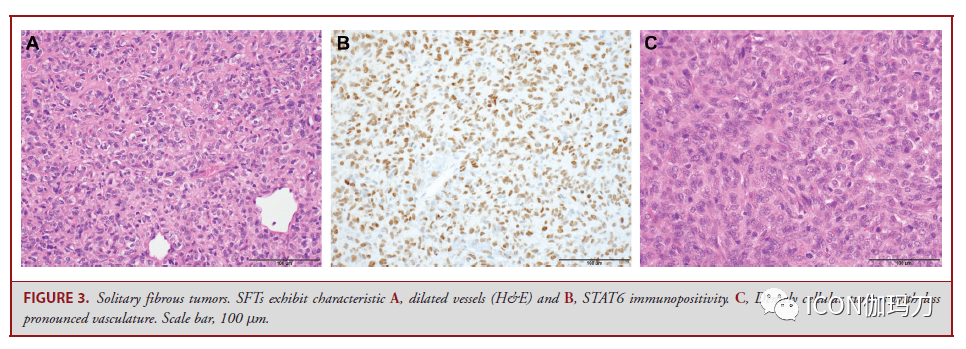
图2。脑膜瘤典型的遗传改动。与脑膜瘤特定亚型相关的突变(注释在括号中),以及与高级别行为相关的特征性染色体改动,可依据脑膜瘤级别分层。 孤立性纤维瘤 孤立性纤维瘤(SFTs)一种间叶成纤维母细胞瘤,常累及颅底,组织学范围从细胞少的到细胞密集的(图3)。细胞少的肿瘤(Hypocellular tumors)梭形-卵形细胞随意排列伴“鹿角形”薄壁扩张的和在胶原基质内的血管。细胞密集的肿瘤的细胞呈圆形-卵圆形,胶原很少,少有明显的血管结构。 孤立性纤维瘤(SFTs)分为3级,按有丝团结活性和存在的坏死来分辨,并与预后相关。1级SFTs是由细胞少的组织学定义的,每10倍的高倍镜视野(HPF)下有丝团结少于5个;2级SFTs为≧5个有丝团结/10HPF,无坏死;3级SFTs为≧5个有丝团结/10HPF伴坏死。在一个系列研讨中,切除范围、肿瘤分级和有丝团结计数被发现是无停顿生存的独立预后标记。 过去,密集细胞肿瘤被称为血管外皮细胞瘤,但往常被了解为SFT的一个组织学谱的一端,基于染色体12q13的单个共享基因组倒置,招致NAB2和STAT6基因两者之间的融合。融合产物的核定位能够经过STAT6免疫反响性来反映,从而将SFT与组织学相似物分辨开来。
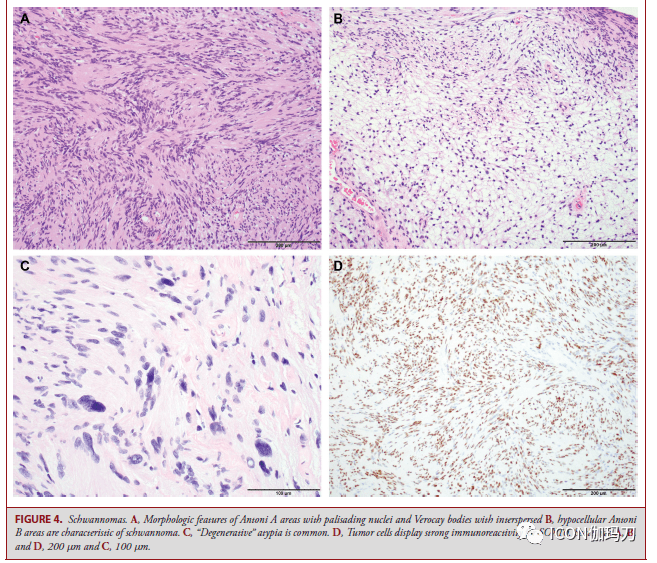
图3。孤立性纤维瘤。SFTs表示为特征A,血管扩张(H&E)和B, STAT6免疫阳性。C,血管系统不明显的细胞密集肿瘤。比例尺,100 μm。 神经鞘瘤 神经鞘瘤是WHO 1级周围神经系统梭形细胞肿瘤。颅内神经鞘瘤最常来源于前庭耳蜗(第八颅)神经的前庭上或下分支。神经鞘瘤的细胞Antoni A区有栅栏状的细胞核和Verocay小体,并散布着细胞少的Antoni B区(图4A和4B)。退行性异型性(图4C)和血管周围透明样变(老化的改动)能够很明显,但与进袭性临床行为无关。囊的周神经细胞表白EMA,肿瘤细胞表白S100和SOX10(图4D)。
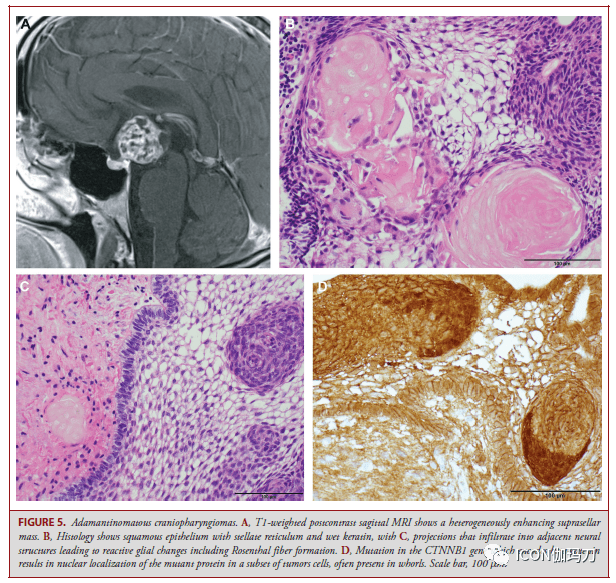
图4。神经鞘瘤。A, Antoni A区呈栅栏状核,Verocay小体散布;B,细胞少的Antoni B区是神经鞘瘤的特征。C、变性异型是常见的。D,肿瘤细胞对SOX10表示出很强的免疫反响性。比例尺A、B、D分别为200 μm和100 μm。 神经鞘瘤可发作于如神经纤维瘤病2型(NF)2和神经鞘瘤病(schwannomatosis)等家族综合征。前庭神经鞘瘤是由NF2的22q12.2号染色体上种系突变招致的NF2肿瘤易理性综合征的典型特征;三分之一或更多的NF2患者发作于无既往家族史的重生突变。其中的双侧前庭神经鞘瘤可作为主要诊断规范。一些契合临床诊断规范的患者是NF2嵌合体,并在发育后期取得躯体NF2突变,因而,缺乏通常经过血液DNA测序检测到的种系NF2突变。发作于非前庭神经(如三叉神经和动眼神经)的神经鞘瘤在非综合征患者中很少见,但在NF2患者中比较常见。多发性肿瘤、年岁轻、沿神经节段不连续或多结节生长,以及丛状散布,提示NF2相关神经鞘瘤。 神经鞘瘤病是由位于22q11染色体上SMARCB1或LZTR1的种系突变以及NF2基因的体细胞(非种系)突变招致的。一个多步骤模型解释了神经鞘瘤病的肿瘤发作,(i)由SMARCB1或LZTR1的遗传种系突变起始,随后(ii) 22号染色体第二副本的丧失(招致NF2的一个副本和SMARCB1和/或LZTR1的第二副本失活)。最后是(iii) NF2的第二副本的体细胞突变,神经鞘瘤病的特征是多发性脊柱的、皮肤的和颅神经鞘瘤。神经鞘瘤病常伴有神经鞘瘤和神经纤维瘤的混合特征,常被误诊为神经纤维瘤或恶性周围神经鞘瘤。单侧前庭神经鞘瘤可作为伴有LZTR1突变的神经鞘瘤病的一部分呈现,该突变在临床上与NF2堆叠,此外,嵌合型NF2与神经鞘瘤病基本堆叠,招致10%的神经鞘瘤病误诊。与NF2不同,神经鞘瘤病的肿瘤与严重和慢性疼痛相关。周围神经鞘瘤和NF2相关的前庭神经鞘瘤在肿瘤细胞间显现洋溢性核SMARCB1 (INI1)免疫反响,而神经鞘瘤病中产生的神经鞘瘤以及NF2相关的非前庭神经鞘瘤显现SMARCB1 (INI1)免疫反响嵌合方式。 对分发神经鞘瘤的基因组研讨显现,在10%的患者中,由于10q染色体上均衡的染色体倒置,存在NF2失活,但也存在染色质调理因子(如ARID1A, ARID1B)和SH3PXD2AHTRA1融合的重复突变。 鞍区病变 颅咽管瘤 颅咽管瘤分为造釉细胞型和乳头状瘤型(adamantinomatous and papillary types),它们具有不同的组织学、遗传驱动和转录组(distinct histologies, genetic drivers, and tranomes)。 造釉细胞型颅咽管瘤 造釉细胞型颅咽管瘤(ACPs)是一种典型的混合实体和囊性肿瘤,在儿童和成人中均能够遇到,来源于漏斗部灰结节轴(infundibulotuberal axis)的任何中央(图5)。绝大多数ACPs在一定水平上累及鞍上区,常累及下丘脑、视觉传导束(visual tract)和神经血管结构。 ACPs组织学表示为低级别鳞状上皮伴星形网状组织和湿角蛋白(图5B);但是,它们经常显现手指状突起浸润到相邻的神经结构,招致反响性胶质改动(图5C)。普遍的组织学方式包含带状生长。恶性转变是十分稀有的,并与预后差相关联,通常粗始起病表示后发作良好并给予放疗(generally occurring well after the primary presentation and the administration of radiation treatment)。 简直一切的ACPs都存在CTNNB1外显子3突变,CTNNB1是编码WNT信号通路调理因子β-catenin的基因。CTNNB1突变招致β-连环蛋白核定位;但是,值得留意的是,固然CTNNB1突变是克隆性的,但仅在肿瘤上皮细胞的一小部分细胞中可见核定位,通常发作在肿瘤细胞的涡漩区(whorls of tumor cells)(图5D),但也存在于分散的簇状和单个肿瘤细胞中。 完整手术切除ACPs可治愈,但常不如乳头状瘤型颅咽管瘤(PCPs)。在复发背景下,,特别是在次全切除后,辅助放射治疗可提供长期的疾病控制。鉴于ACPs在儿童中占主导位置,在对这些患者的长期管理中,必须思索与疾病及其治疗相关的生活质量和长期并发症发作率,包含下丘脑瘦削、认知后遗症和代谢紊乱。
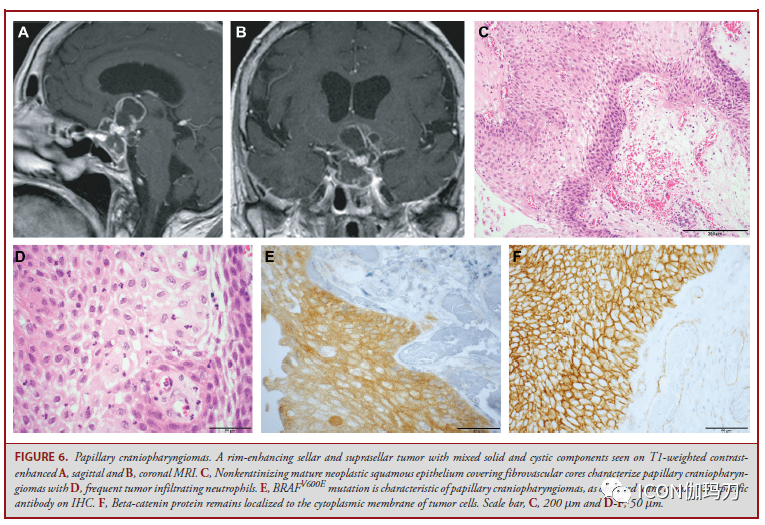
图5.成釉细胞型颅咽管瘤。A,T1加权增强后矢状位MRI显现鞍上肿块不平均强化。B,组织学显现鳞状上皮伴星形网状和湿角蛋白,C,突出浸润到临近神经结构招致反响性胶质改动,包含Rosenthal纤维构成。D,编码-连环蛋白的CTNNB1基因突变招致肿瘤细胞亚群中突变蛋白的核定位,通常呈涡漩状。比例尺,100 μm。 乳头状瘤型颅咽管瘤 与ACP相比,乳头状瘤型颅咽管瘤(PCP)主要发作在成人,很少发作在儿科患者。PCPs也可发作在沿颅咽管的任何中央,但更倾向于位于第三脑室底的漏斗部和灰结节质,较少位于鞍区间隙(图6A和6B)。一些PCP 扩展到第三脑室腔,而另一些则出往常完好的脑室底之上,完整位于脑室自身。 PCP通常表示出共同的组织学特征,包含掩盖纤维血管中心或囊肿壁的非角化成熟肿瘤鳞状上皮(图6C)。肿瘤浸润中性粒细胞多见,常可见细胞间桥(图6D)。星形网状组织和湿角蛋白缺失,钙化稀有。PCP患者的肿瘤-脑界面通常界线分明,无侵袭性突起,因而PCP患者比ACP患者更容易完整切除。 大多数PCP患者在BRAFV600E中表白典型突变,可经过特异性抗体辨认(图6E)。在诊断理论中,重要的是要留意有大的BRAFV600E抗体,伴在纤毛中的基因丝动力蛋白(axonema dynein proteins in cilia),使对Rathke裂囊肿(RCC)的评价复杂化,特别是对鉴别PCP重要的伴普遍鳞状上皮化生的RCC。此外,PCP中,位于细胞质膜的β-连环蛋白(图6F),而不是像在ACP中那样位于细胞核上(图5D)。 在一些研讨中提出PCP预后较好,但在其他研讨中未得到证明。PCP的初级治疗是手术,必要时辅以放疗;但是,在简直一切PCP中发现BRAFV600E突变为患者管理提供了新的治疗机遇。在新诊断和复发的BRAFV600E突变PCP患者中,运用BRAF和/或促丝裂原活化蛋白激酶(MEK)抑止剂治疗的患者中,已显现出快速和显著的治疗反响。添加MEK抑止剂是由于它们已被证明能够减少黑色素瘤耐药的呈现,以及与BRAF抑止剂单药治疗相关的鳞状细胞癌等并发症的展开。但是,在中止BRAF抑止剂达拉非尼(dabrafenib)治疗后,察看到疾病得到了耐久的控制的多中心2期临床实验分离抑止BRAF和MEK的维莫非尼( vemurafenib)和考比替尼(cobimetinib )(NCT03224767)曾经完成了招募,且全部结果估量在2021年晚些时分展示维莫非尼( vemurafenib)/考比替尼(cobimetinib )招致在一切接受1或多个周期治疗的患者有客观反响。
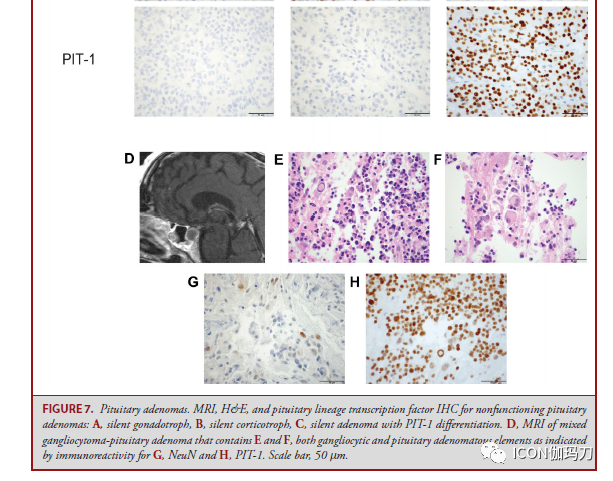
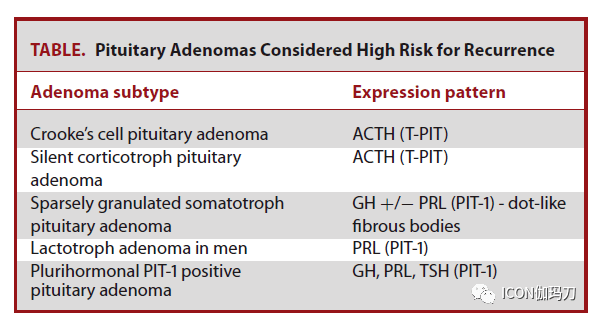
图6.乳头状瘤型颅咽管瘤。A,矢状位和B冠状位MRIT1加权增强扫描可见鞍区和鞍上边沿强化的肿瘤,混合有实质性和囊性成分。C,非角化的成熟肿瘤鳞状上皮掩盖纤维血管中心,为乳头状瘤型颅咽管瘤的特性;d,肿瘤经常浸润中性粒细胞。E,在免疫组化上运用突变特异性抗体察看到 ,BRAFV600E突变是乳头状瘤型颅咽管瘤的特征。F, β -连环蛋白依旧位于肿瘤细胞的细胞质膜。比例尺,C,200 μm;D-F,50 μm。 垂体腺瘤/垂体神经内分泌肿瘤 垂体腺瘤是发作在腺垂体(或很少发作在临近异位)的肿瘤性增生,有时也被特指(designated)是垂体神经内分泌肿瘤。垂体肿瘤有多种分类措施。垂体腺瘤局限于鞍区和鞍旁区域,而有颅脑脊髓或全身扩散的则被归类为垂体癌。分泌一项或多项垂体腺瘤激素,如泌乳素(PRL)、生长激素(GH)、促肾上腺皮质激素(ACTH)、促甲状腺素(β-TSH)、促黄体生成素(β- LH)和卵泡刺激素(βFSH),被以为是“功用性”的,而那些不分泌激素的则被以为是“无功用性的”。 在过去的十年中,垂体腺瘤的分类阅历了实质性的修订,从仅仅依据激素的产生来定义腺瘤,转为整合关于垂体腺垂体细胞谱系的额外信息。评价垂体激素的表白依旧是这些肿瘤分类的请求,功用性腺瘤与肿瘤激素表白的相关性是必要的。但是,以前,缺乏免疫组化激素表白的肿瘤被称为“零细胞腺瘤”;往常,绝大多数(约95%)的这些肿瘤被发现表明分化为特定的激素谱系的表白3个转录因子中的1个(SF-1, T-PIT, PIT-1)。超越三分之二以前认定为零细胞腺瘤的,基于SF-1的表白,往常被以为是静默性促性腺激素细胞垂体腺瘤(图7A),另外四分之一是表白T-PIT的静默性促肾上腺皮质激素细胞垂体腺瘤(图7B),其他为表白PIT-1的静默性生长激素细胞、泌乳素细胞或促甲状腺素细胞腺瘤(图7C)。缺失一切3个转录因子(SF-1, T-PIT, PIT-1)和任何激素的表白往常定义为零细胞腺瘤。某些组织学亚型与较具进袭性行为和有较大的复发倾向相关(表)。当遇到缺乏谱系转录因子表白的肿瘤时,应积极思索腺瘤以外的实体,如神经垂体肿瘤(即垂体细胞瘤、颗粒细胞瘤、梭形细胞嗜酸细胞瘤),或被血液癌累及,或因肺癌、乳腺爱和前列腺癌扩散转移。
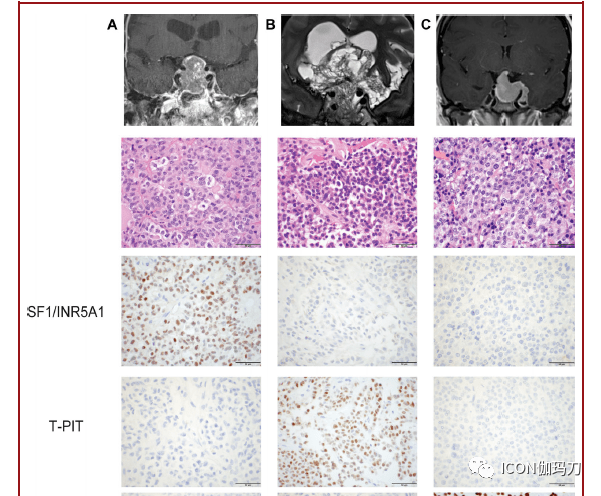
图7。垂体腺瘤。无功用垂体腺瘤的MRI、H&E和垂体谱系转录因子IHC: A,静默性促性腺激素细胞,B,静默性促肾上腺皮质激素,C,静默性PIT-1分化腺瘤。D,包含E和F的混合神经节细胞瘤-垂体腺瘤的MR,表示为神经节细胞和垂体腺瘤成分,与G有免疫反响性;NeuN和H, PIT-1的比例尺,50 μm。 表:被以为有高复发风险的垂体腺瘤。 腺瘤亚型 表白方式 Crooke 细胞腺瘤 ACTH(T-pit) 静默性促肾上腺皮质激素细胞腺瘤 ACTH(T-pit) 稠密颗粒性生长激素细胞垂体腺瘤 GH+/-PRL(PIT-1)点状纤维小体 男性泌乳素细胞腺瘤 PRL(PIT-1) 多激素PIT-1阳性垂体腺瘤 GH,PRL,TSH(PIT-1)
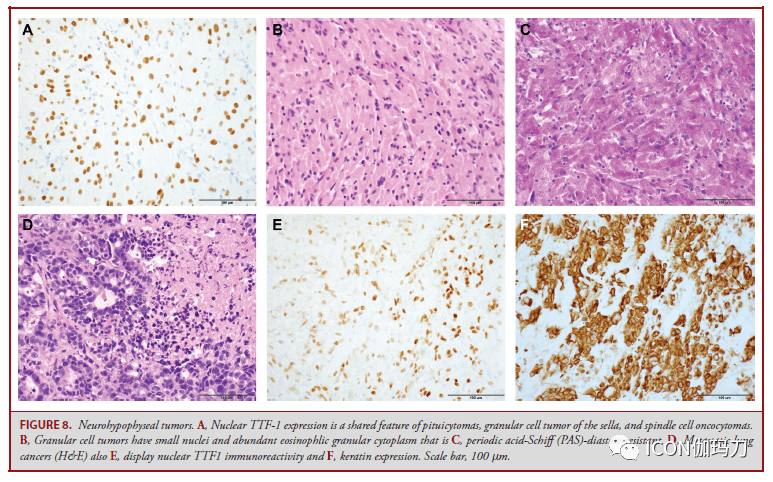
固然能够应用谱系转录因子对垂体腺瘤中止分类,但常常经过火子措施检测到激素谱系的转分化。很少状况下,垂体腺瘤的转分化在组织学上很明显,肿瘤表示为神经节细胞和垂体腺瘤成分,称为混合性神经节细胞瘤垂体腺瘤(图7D-7H)。转分化肿瘤患者发作内分泌疾病,最常见的是肢端肥大症和高泌乳素血症。 GNAS基因突变是垂体腺瘤中常见的复发性遗传异常,在40%的生长激素细胞腺瘤中发作,由于环状腺苷一磷酸蛋白激酶A通路的激活招致生长激素超高分泌。 在分泌ACTH的和分泌GH的腺瘤中曾经发现了激活突变。去泛素酶基因USP8和USP48的突变共占库欣病患者的近三分之二,招致促肾上腺皮质激素的前体阿黑皮素原 (POMC)增加。此外,16%的促肾上腺皮质激素细胞腺瘤中察看到BRAFV600E突变。表观遗传改动和染色体不稳定可能进一步有助于垂体腺瘤的发病机制,特别是在功用性腺瘤或静默性促肾上腺皮质激素细胞腺瘤。一小部分垂体腺瘤发作在遗传性肿瘤易感综合征中,最常见的是AIP(家族孤立性垂体腺瘤综合征)、GPR101 (X连锁肢端肥大伟人症)、MEN1、PRKAR1A (Carney 复合征)、SDHx(伴嗜铬细胞瘤和副神经节瘤)。 垂体细胞瘤,鞍区颗粒细胞瘤,梭形细胞嗜酸细胞瘤 垂体细胞瘤、鞍区颗粒细胞瘤和梭形细胞嗜酸细胞瘤是一个稀有的、典型的良性、生长迟缓的肿瘤家族,发作于中年人,由漏斗部和垂体后叶的垂体细胞惹起(WHO分级1级)。这些肿瘤表示为一个单一疾病实体的组织学和临床谱系,具有形态学堆叠和衔接的核TTF-1表白(图8A)。垂体细胞瘤有细长的双极梭形细胞,呈片状和成簇,偶尔与血管周围的淋巴细胞呈席纹状排列,而颗粒细胞瘤有片状或结节状的多边形细胞、核小、丰厚的过碘酸-希夫(PAS)染色反响阳性和淀粉酶抵御性颗粒状嗜酸性胞浆(图8B和8C),梭形细胞嗜酸性细胞瘤的细胞呈交错成簇的拉长的细胞,嗜酸性细胞特征可变。梭形细胞嗜酸细胞瘤表示出抗线粒体抗体免疫反响性,而颗粒细胞瘤表白α -1抗胰蛋白酶和CD68,并有胞浆内颗粒121淋巴细胞灶性汇集是常见的。大多数病例表示为低增殖和稀有的有丝团结。这些肿瘤缺乏垂体激素、转录因子和神经内分泌标记物的表白。核TTF-1也可在非肿瘤性垂体细胞和转移性肺或甲状腺癌中察看到表白(图8D-8F)。
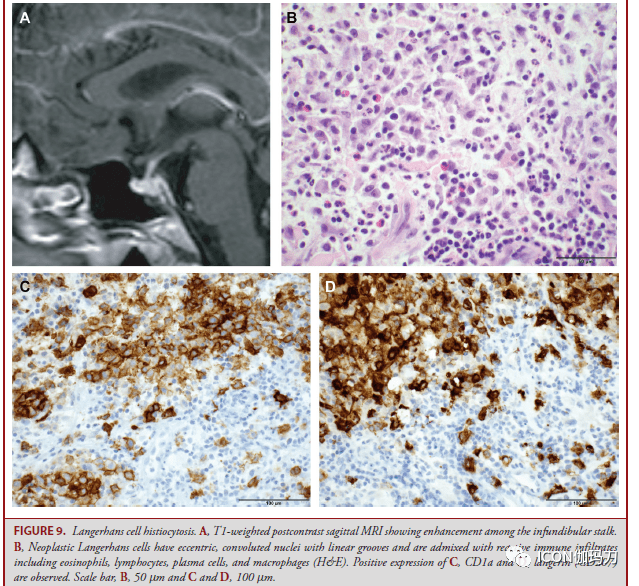
图8。垂体神经部肿瘤。A,核TTF-1表白是垂体细胞瘤、鞍区颗粒细胞瘤和梭形细胞嗜酸细胞瘤的共同特征。B,颗粒细胞肿瘤具有小核和丰厚的嗜酸性颗粒胞浆,C,抗过碘酸-希夫(PAS)淀粉酶。D,转移性肺癌(H&E),E,显现核TTF1免疫反响性;F,角蛋白表白。比例尺,100 μm。 这些低级别肿瘤的预后通常很好,切除后没有复发;但是,梭形细胞嗜酸细胞瘤可表示出较强的进袭性行为,其复发发作率高于垂体腺瘤的。 垂体母细胞瘤 垂体母细胞瘤是2岁以下儿童的稀有肿瘤,通常发作于鞍区,常有鞍上和鞍旁伸展,由3个组织学部分组成:小而未分化(原始)的母细胞,大的神经内分泌细胞排列成小叶或洋溢性片状,以及原始的Rathke型上皮。垂体转录因子表白稀有。患者常因肿瘤细胞产生促肾上腺皮质激素而表示有库欣综合征,并可因视交叉受累而呈现眼肌麻木。简直一切报道的肿瘤都有DICER1基因的种系突变,患者可能展开为其他良性和恶性肿瘤和,如胸膜肺母细胞瘤,与DICER综合征相关的发育异常病变。 朗格汉斯细胞组织细胞增生症 朗格汉斯细胞组织细胞增生症(LCH)是由髓系前体克隆扩增而成,分化为相似于皮肤的朗格汉斯细胞的CD1a+和Langerin/(CD207)+肿瘤细胞,即表皮的初级抗原提呈细胞(the primary antigen presenting cell of the epidermis)。在中枢神经系统,LCH累及颅底或颅骨,可能延伸至周围软组织、脑膜或垂体-下丘脑轴(图9A)。 肿瘤性朗格汉斯细胞的核呈偏心性、缠绕的,有线性凹槽,并与包含嗜酸性粒细胞、淋巴细胞、浆细胞和巨噬细胞等反响性免疫浸润物混合(图9B),表白CD1a(图9C)和/或langerin(CD207)(图9D)。 大约50%的LCH存在BRAFV600E突变,132-134,在既往多次治疗失败的高危LCH患者中,BRAF抑止剂维莫非尼(vemurafenib)有很好的应对;但是,中止治疗后,肿瘤疾速呈现值得留意的是,无论有包含ARAF、BRAF、RAF1、NRAS、KRAS、MEK1和MEK2.肿瘤基因型突变的LCH患者运用MEK抑止剂考比替尼(cobimetinib)都取得了耐久的应对,需求长期有效的维持战略。
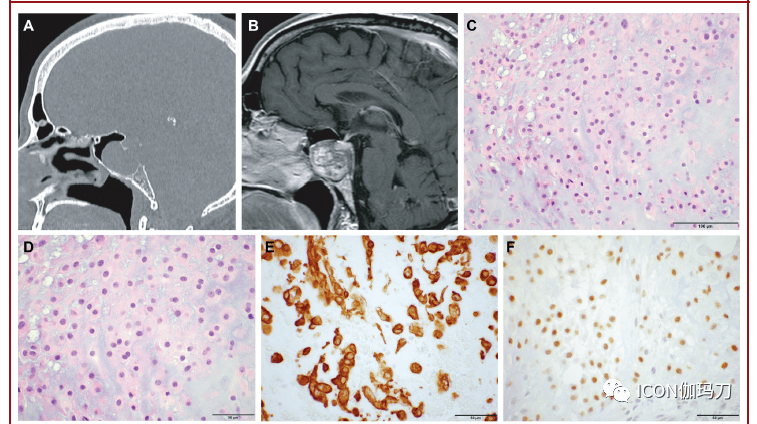
图9。朗格汉斯细胞组织细胞增生症。A,T1加权增强矢状位MRI显现漏斗部垂体柄增强。B,肿瘤性朗格汉斯细胞的核呈偏心性、旋绕的,有线状凹槽,并与反响性免疫浸润物混合,包含嗜酸性粒细胞、淋巴细胞、浆细胞和巨噬细胞(H&E)。C、CD1a、D、langerin (CD207)阳性表白。比例尺:B, 50 μm; C, D, 100 μm。 脊索瘤 脊索瘤是显现脊索分化的恶性肿瘤,位于中线,有骶尾骨区和斜坡夫人倾向(with a propensity for the sacrococcygeal region and the clivus(图10A和10B)。脊索瘤可表示为疼痛或神经病症,招致骨破坏,神禁受压榨,或颅神禁受累。它们包含几种组织病理学类型,包含常规、软骨样、去分化和低分化类型。 与它们的来源分歧,一切脊索瘤都表白由TBXT基因编码的脊索谱系转录因子短链蛋白(brachyury)。四分之一的患者有TBXT基因的重复,而10%至20%的患者有磷酸肌醇-3-激酶(PI3K)信号成分的临床可执行突变(clinically actionable mutations in phosphoinositide-3-kinase (PI3K) signaling components)。 在常规脊索瘤中,肿瘤细胞具有嗜酸性胞浆,常嵌在黏液样基质小叶中,构成粘连的巢状和索状,但也可呈片状或单细胞(图10C和10D)。脊索瘤的鲜明特性是细胞质有液泡伴空泡细胞。软骨样脊索瘤具有相似透明软骨的细胞外基质,可能与软骨肉瘤相混杂,但可经过细胞角蛋白阳性表白和鼠短尾突变体表型表白来分辨(图10E和10F)。去分化脊索瘤具有双相形态,常规和软骨样成分混杂着肉瘤成分;常规成分和软骨样成分表白短链蛋白(brachyury)和细胞角蛋白,而肉瘤成分均不表白。 低分化脊索瘤的特征是INI1蛋白缺失,是22q11染色体上SMARCB1基因的分别缺失的结果。低分化脊索瘤包含梭形或上皮样细胞,保存短链蛋白表白而缺乏INI1。这是脊索瘤的独一亚型,主要发作在儿童,中位年龄为7岁,而其他脊索瘤类型的中位年龄为45 - 60岁。与常规脊索瘤相比,低分化和去分化脊索瘤表示出特别具有进袭性的行为,中位生存期较差。察看到的1p36和9p21染色体缺失是脊索瘤复发的独立预测因子。 软骨肉瘤 软骨肉瘤是伴软骨分化的间叶细胞的恶性肿瘤(mesenchymal malignancies with cartilaginous differentiation),表示为神经系统的受压榨和颅神禁受影响(neural compression and cranial nerve impingement)。颅内软骨肉瘤占一切软骨肉瘤的1%,岩斜交界处是最常见的部位,其次是颞枕交界处(图10G和10H)。存在几种亚型,包含常规、去分化和透明细胞软骨肉瘤。依据异型性水平、细胞多样性和有丝团结活性将肿瘤分为3级,1级为分化良好,2级为中分化,3级为低分化。颅底软骨肉瘤最常见的是1级或2级和常规亚型。 透明样常规软骨肉瘤由腔隙内(lacunar spaces)呈固体嗜碱性基质(solid basophilic matrix)的肿瘤细胞组成,(图10I和10J),而黏液样常规软骨肉瘤在黏液基质中呈梭形和星形肿瘤细胞。脊索瘤中粘连细胞缺失。肿瘤细胞对S100有免疫反响(图10K),但细胞角蛋白和短链蛋白呈阴性(图10L)。
图10。脊索瘤和软骨肉瘤。A,在矢状面CT上扩张性肿物累及上部斜坡并延伸至蝶窦,招致下方骨溶解性腐蚀。B, 增强后矢状面MRI T1加权上的不平均强化。C和D,脊索瘤细胞内嵌于黏液样基质小叶中的粘连巢和索。脊索瘤的病理表示为E、细胞角蛋白和F的阳性表白。G,冠状CT显现左侧岩斜交界处呈现密集钙化的轴外外生性肿块,H, T1增强冠状MRI上不平均强化,下方脑实质受压。I和J,在固体嗜碱性基质中腔隙内的肿瘤细胞标记着透明样常规软骨肉瘤。软骨肉瘤的诊断进一步得到K,S100免疫反响阳性,和L,缺乏短链蛋白(brachyury)的表白,的支持。比例尺,D-F和J-L, 50 μm, C, 100 μm, I, 200 μm。 超越85%的颅底软骨肉瘤存在IDH1突变,而颅内软骨肉瘤中最常见的突变是IDH1R132C,而胶质瘤中IDH1R132H占优势。固然大多数软骨肉瘤是分发性的,但由Ollier病和Maffucci综合征惹起的内生软骨瘤病(enchondromatosis)患者展开为软骨肉瘤的风险增加。包含低级别胶质瘤的许多其他恶性肿瘤与这些病症有关。 副神经节瘤 副神经节瘤(Paragangliomas)是由来源于特别的神经嵴细胞的神经内分泌肿瘤。脊髓副神经节瘤是硬膜内的,通常来源于马尾终丝,偶尔也来源于尾神经根。颈部和胸部位置远不常见。颅内副神经节瘤通常是由于颈静脉球副神经节瘤延伸至颅骨所致,有30%以上的副神经节瘤发作在此部位(图11A)。但在其他部位中,已报道在桥小脑角(CPA)、蝶鞍、和小脑的确发作完整颅内的副神经节瘤。颈静脉球副神经节瘤延伸至颅骨的患者常表示为后组颅神经功用障碍和搏动性耳鸣。 副神经节瘤是界线分明的(well-demarcated)和分化分化肿瘤大小平均的圆形或多边形主(I型)细胞与圆形和卵圆型核出往常称为Zellballen的小叶和巢中,被单层纺锤形支撑(II型)细胞包抄的和毛细血管和一个懦弱的支持网硬蛋白纤维网(图11B和11C)。主细胞显现出嗜铬粒蛋白A和突触素的强免疫反响(图11D)。S-100在主细胞中有不同的表白,但在支持细胞中有标记(图11E)。对这些有血管蒂的肿瘤中止动脉供血的栓塞能够减少切除过程中的失血(图11F)。 大约三分之一来源自成人的副神经节瘤是遗传性的。超越四分之三的肿瘤是由RET (MEN 2)、VHL (von Hippel-Lindau)、NF1或SDH基因之一(SDHA、SDHB、SDHC、SDHD、SDHAF2)的突变惹起的。其他突变基因包含EGLN1、EGLN2、EPAS1、FH、KIF1B、MAX、MDH2、MEN1和TMEM127。综合征性肿瘤发作于较年轻的年龄,通常是多发性的。大约10%的分发性肿瘤患者(无家族史或综合征表示)被发现有遗传性疾病。
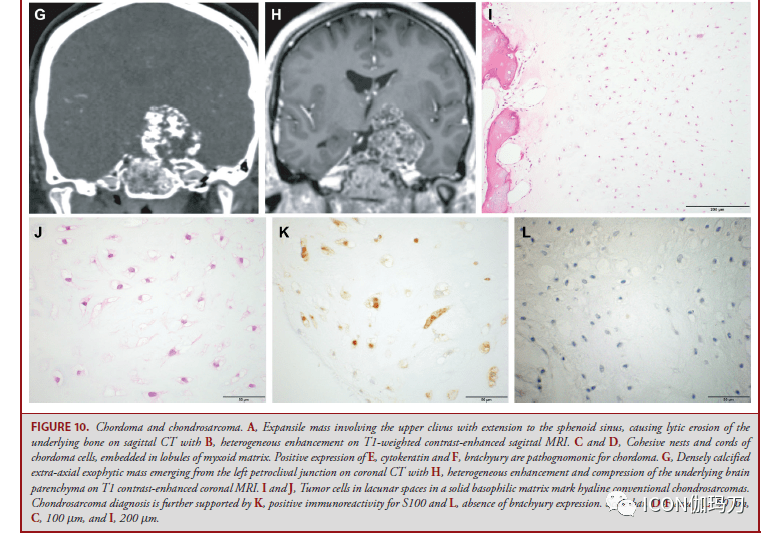
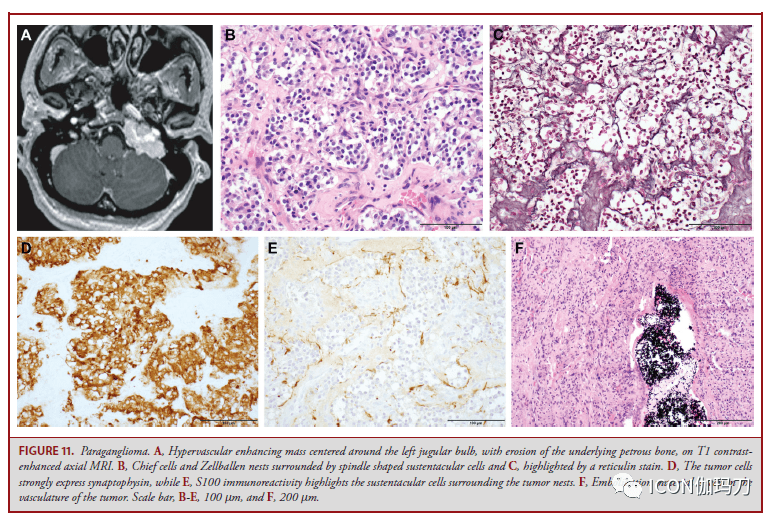
图11。副神经节瘤。A,轴位MRI T1增强扫描显现,以左颈静脉球为中心的富血管强化肿块,伴有岩骨下腐蚀。B,主细胞和Zellballen巢被梭形支持细胞包抄;C,用网状染色突出显现。D,肿瘤细胞激烈表白突触素,而E, S100免疫反响突出了肿瘤巢周围的支持细胞。肿瘤血管内可见栓塞物质。比例尺,B-E, 100 μm, F, 200 μm。 内淋巴囊肿瘤 内淋巴囊肿瘤(ENDOLYMPHATIC SAC TUMOR ,ESLT)是生长迟缓,部分进袭性乳头状上皮肿瘤,来源于内淋巴囊和导管的上皮。ESLTs发作在10%到20%的VHL肿瘤易感综合征的患者中。分发性ESLT是稀有的。早期病症包含感音性神经性耳聋、耳鸣和眩晕,晚期病症包含由于CPA扩散而累及面部和前庭耳蜗神经的神经损伤。影像学典型显现内淋巴囊后部岩骨腐蚀(图12A和12B)。肿瘤组织学上相似脉络膜丛乳头状瘤(图12C和12D),表白细胞角蛋白(图12E),但可经过核顶端定位和骨质受累来鉴别。
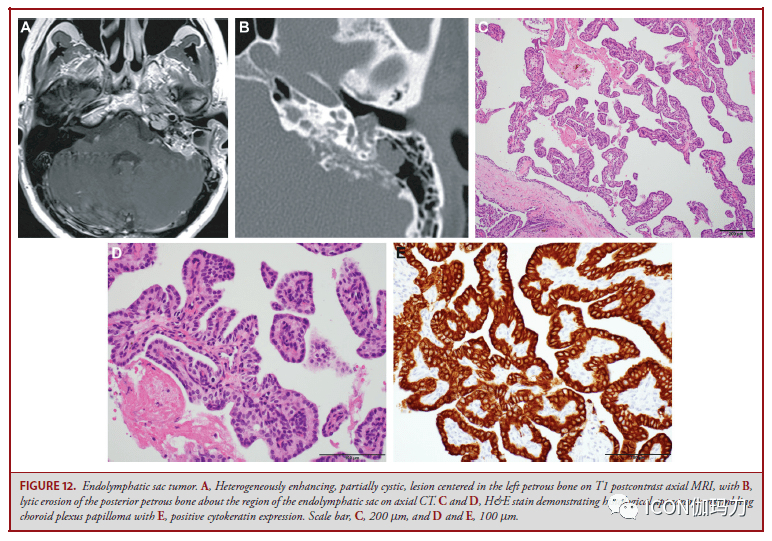
图12。内淋巴囊肿瘤。A,轴位MRI T1增强,不平均强化,部分囊性,病变中心位于左侧岩骨,B,轴位CT示内淋巴囊区域后岩骨溶解性腐蚀。C和D, H&E染色显现相似脉络膜丛乳头状瘤的组织学外观,E,细胞角蛋白阳性表白。比例尺C 200 μm, D和E 100 μm。 对未来的见地 颅底肿瘤的诊断和治疗正阅历着分子时期的转变。相似于神经胶质瘤和全身系统性癌症的基因组分类的影响,颅底肿瘤越来越被以为具有典型的基因改动,这反过来影响和反映其表型和临床结果。 针对特定基因变异的药物临床实验(即基因驱动的临床实验)曾经被开发出来用于一些颅底肿瘤;这些措施的有效性将在未来数月和数年讲演,且继续细化优势。颅底肿瘤的分子改动进一步反映了它们的胚胎来源,并允许对传统组织病理学中止无成见方式的分类。肿瘤表观遗传学的研讨有望进一步明白疾病类别,从而促进诊断和预后。由于颅底肿瘤的稀有性,深化了解其发病机制需求跨多个中心中止前瞻性研讨。对颅底肿瘤生物机制的进一步了解将为新的治疗战略提供信息,并指导在这些具有解剖学应战性的肿瘤中明智地应用手术和放射治疗的机遇和原理。 神外前沿 E-mail:shenwaiqianyan@qq.com; |

 万奢网手机版
万奢网手机版
官网微博:万奢网服务平台